The current coronavirus (COVID-19) pandemic is an unprecedented public health emergency in the U.S. Even though medical professionals are working around the clock to conduct testing, the problem still remains to be a shortage of diagnostic kits. Aimed to improve the limited diagnostic capability and to fight against the COVID-19 public health crisis, the FDA issued several emergency policies since late February.
On February 4th, the U.S. Food and Drug Administration (FDA) issued the first Emergency Use Authorization (EUA) to the 2019-nCoV RT-PCR Diagnostic Panel developed by the U.S. Centers for Disease Control and Prevention (CDC). In the meantime, FDA was actively collaborating with other manufacturers and developers of COVID-19 diagnostic kits.
Due to the increase in demand, on February 29th, FDA issued an emergency policy to help accelerate the speed at which laboratory and commercially developed COVID-19 diagnostic tests can be put to use. Between then and March 16th, only 4 commercial kits have been granted EUA by the FDA and could be immediately used in SARS-CoV-2 viral sequence diagnostics testing in labs certified under the Clinical Laboratory Improvement Amendments (CLIA).
However, the rapid and unpredictable development of this new public health crisis is unprecedented. Hence, in order to further expand testing capabilities in the U.S., on March 16 the FDA updated the policy issued on Feb. 29, providing more regulatory relief during the coronavirus outbreak in the U.S. In chapter IV, part C, the guidance addressed that “FDA does not intend to object to a commercial manufacturer’s development and distribution of SARS-CoV-2 test kits for specimen testing for a reasonable period of time after the manufacturer’s validation of the test…” As clarified later in the statement, "a reasonable period of time" is defined as 15 days. This means, commercial manufacturers are able to distribute newly developed COVID-19 diagnostic kits whose “performance characteristics have already been validated by the manufacturer” as early as 15 days prior to submitting the EUA application to the FDA.
Click to download FDA latest policy
The newly updated policy significantly accelerated the speed at which kits developed by commercial manufacturers – who are usually better equipped to rapidly manufacture these kits on a large scale – become available for use. This includes the one ABclonal is currently distributing in the U.S. – 3DMed’s ANDiS SARS-CoV-2 RT-qPCR Detection Kit. The kit’s EUA application was received by FDA on March 4th. Since then, it has passed two rounds of FDA review and is currently awaiting final approval. Based on the new policy, the detection kit is qualified for use in the in vitro diagnostics of COVID-19.
3DMed’s EAU application waiting for final review

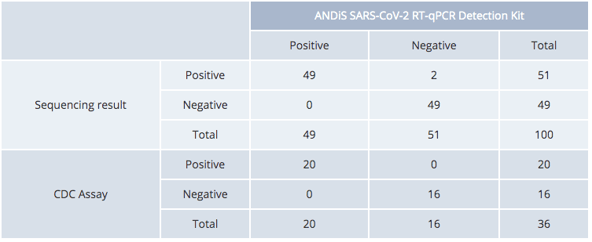
The 3DMed SARS-CoV-2 RT-qPCR detection kit have shown 100% specificity and 99.3% sensitivity in a clinical trial consisting of 100,000 samples. Additional test results demonstrate a 98% agreement with sequencing-based test results and 100% agreement with tests developed by the Centers for Disease Control and Prevention (CDC).
3DMed Kit vs NGS/CDC Assay

To help increase testing efficiency, ABclonal is also distributing 3DMed’s ANDiS 350 Automated Nucleic Acid Extraction System, a machine also accepted for review by the FDA. When used with the ANDiS Viral Nucleic RNA Auto Extraction & Purification Kit by 3DMed, the machine can increase daily throughput by 5 to 25 times compared to traditional manual extraction, significantly improving diagnostic capability with limited lab workers to meet the current escalating COVID-19 diagnostic demand.
 |
 |


If you have any question related to products or want to order in bulk, please call us at 888-754-5670 or email us at service@abconal.com for detailed information. We will respond you as soon as possible.
Resouces:
[1] https://www.fda.gov/emergency-preparedness-and-response/mcm-issues/coronavirus-disease-2019-covid-19
[2] https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-diagnostic-tests-coronavirus-disease-2019-during-public-health-emergency